Target-aware drug discovery has greatly accelerated the drug discovery process to design small-molecule ligands with high binding affinity to disease-related protein targets. Conditioned on targeted proteins, previous works utilize various kinds of deep generative models and have shown great potential in generating molecules with strong protein-ligand binding interactions. However, beyond binding affinity, effective drug molecules must manifest other essential properties such as high drug-likeness, which are not explicitly addressed by current target-aware generative methods. In this article, aiming to bridge the gap of multi-objective target-aware molecule generation in the field of deep learning-based drug discovery, we propose ParetoDrug, a Pareto Monte Carlo Tree Search (MCTS) generation algorithm. ParetoDrug searches molecules on the Pareto Front in chemical space using MCTS to enable synchronous optimization of multiple properties. Specifically, ParetoDrug utilizes pretrained atom-by-atom autoregressive generative models for the exploration guidance to desired molecules during MCTS searching. Besides, when selecting the next atom symbol, a scheme named ParetoPUCT is proposed to balance exploration and exploitation. Benchmark experiments and case studies demonstrate that ParetoDrug is highly effective in traversing the large and complex chemical space to discover novel compounds with satisfactory binding affinities and drug-like properties for various multi-objective target-aware drug discovery tasks.
Introduction
The rational design of molecules to act as clinical drugs remains a significant challenge in biopharmaceutical research, especially concerning the attainment of favorable physiochemical and pharmacological properties. In support of such endeavors, target-based drug discovery aims to identify small-molecule ligands that exhibit high affinity and specificity for a particular protein pocket structure1. Traditionally, target-based drug discovery has been approached through either high-throughput experimental methods or virtual screening of extensive chemical databases2,3 targeted at specific biomolecular targets4,5. Subsequently, the screening of bioanalytical indicators through elaborate clinical experiments is conducted to evaluate drug-like properties. This pursuit contributes to the conventional 10-year drug development cycle and staggering research and development costs of approximately 2.8 billion USD, coupled with a remarkably high failure rate. The predetermined selection of compounds for screening further constrains the exploration of chemical space, tethering it to historical knowledge derived from previously investigated molecules. This ultimately leads to a fervent industry focus on popular drug targets, resulting in the challenge that the molecules selected through screening are unable to avoid patent restrictions. In contrast, recent advancements in target-aware molecule generation, particularly the development of generative models trained on extensive datasets, present a promising paradigm shift. These models, rooted in deep learning, offer an innovative approach to expedite ligand discovery and optimization. They achieve this by generating entirely novel and diverse molecules capable of binding to a specified protein target, starting from scratch6. This transformative approach holds great potential to overcome the limitations associated with traditional methods, offering a more efficient and expansive exploration of the entire chemical space.
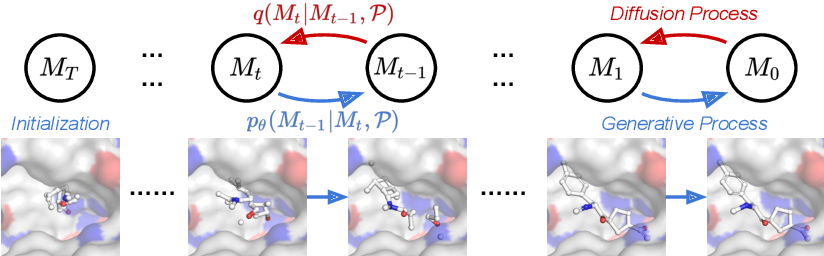
Since the first inception of an autoencoder model conditioned on targeted proteins in 20187, there has been rapid progress in deep learning-based target-aware molecule generation methods. Various works take advantage of conditional generative models, such as the autoencoder7,8,9, generative adversarial network10, and diffusion model11,12, to infer entire molecules through a one-time feedforward process, incorporating binding site information as input. Moreover, to enhance structural representation, convolutional neural networks7 and graph convolutional networks8 are employed. In the meantime, some approaches utilize voxelized representations10 or atomic density grids13 to characterize compound-receptor complexes. Another pivotal category of deep learning-based target-aware drug discovery involves autoregressive generative models, which predict the next atom (and its position) sequentially conditioned on the molecular fragment and binding site information. To model the conditioned intermediate context, diverse network architectures like transformers14,15, recurrent neural networks16,17, and flow models18 are introduced as the context encoder. Additionally, graph neural networks18,19,20 are widely utilized to extract chemical and geometrical features of ligands and protein pockets. However, these efforts are not yet integrated into mainstream drug discovery practices, and a significant obstacle lies in the inherent multi-objective optimization nature of drug discovery 21. Beyond strong binding affinity to the targeted protein, drug molecules must exhibit other desirable properties, such as high drug-likeness and low toxicity. Presently, existing deep learning-based target-aware generative methods predominantly focus on the single objective of optimizing binding affinity. The multi-objective nature of drug molecules, with sometimes conflicting demands, necessitates ongoing development of novel multi-objective target-aware drug discovery techniques to enhance the overall success rates of drug discovery.
Conversely, numerous studies have explored the domain of general multi-objective drug discovery. Certain approaches, such as MolGPT22, fall within the ligand-based methodology, aspiring to generate novel compounds with favorable physicochemical properties. However, these methods fall short in incorporating protein information, thus lacking assurance that the generated molecules can effectively bind to specified protein targets. Concurrently, other methodologies like MCMG21, RationaleRL23, MolSearch24, and GENERA25 aim to optimize not only the binding affinity objective but also other property objectives. Specifically, these methodologies leverage optimization techniques such as reinforcement learning26 and genetic algorithms27 to enhance the binding affinity objective predicted by machine learning-based or simulation-based docking score functions. However, a notable drawback is their failure to explicitly incorporate target protein information when constructing generative models. The absence of protein information renders the optimization of the binding affinity objective inefficient, and the resulting generative models from these target-scoring-based methods cannot be readily generalized to other protein targets. In contrast to ligand-based and target-scoring-based approaches, a recent development is CProMG28, designed to generate molecules that meet multiple property constraints with an enhanced representation of protein structure information. CProMG treats this task as a multi-constraint molecule generation problem, with each property constraint set to exceed a predefined threshold. However, CProMG does not attempt to maximize molecule properties through optimization techniques for a comprehensive exploration of the chemical space. A more in-depth discussion is provided in the Discussion section.
Similar challenges also exist in natural language generation tasks, where models predicting the next token often express unintended behaviors, such as making up facts, generating biased or toxic text, or not following user instructions. To address this issue, OpenAI focuses on fine-tuning approaches to align language models. Specifically, they employ reinforcement learning from human feedback (RLHF) to fine-tune GPT-329 to follow a broad class of written instructions30. In contrast to the fuzzy, hard-to-quantify human values in natural language tasks, we can explicitly calculate multiple molecular metrics in the context of drug development.
In this study, we explore the use of an autoregressive Pareto Monte Carlo Tree Search (MCTS) generation algorithm named ParetoDrug for the design of drug molecules to address the existing gap in multi-objective target-aware drug discovery within the domain of deep learning-based drug discovery. This algorithm effectively facilitates the simultaneous optimization of multiple molecule properties. In its operation, ParetoDrug first explores molecules on the Pareto Front within the chemical space. It achieves this by maintaining a global pool comprising Pareto optimal molecules, each of which is not surpassed by another molecule in the same pool across every property objective. During the exploration process, ParetoDrug leverages existing pretrained autoregressive target-aware molecule generation models to guide the search for the next atom symbol, facilitating the identification of molecules with high binding affinity to protein targets. Additionally, in the selection of the next atom symbol, ParetoDrug introduces a scheme named ParetoPUCT. This scheme is designed to balance the exploration of chemical space and the exploitation of the pretrained autoregressive generative model. Through these strategies, ParetoDrug owns the ability to generate molecules with multiple desirable properties, including binding affinity. Computational evaluations on the benchmark dataset and case studies, including multi-objective target-aware drug discovery tasks for known drugs (e.g., Tropifexor and Copanlisib), a multi-target drug discovery task for HIV-related disease targets, and a multi-target multi-objective drug discovery task for a dual-inhibitor Lapatinib, demonstrate the high effectiveness of ParetoDrug. The algorithm exhibits proficiency in discovering small-molecule drug candidates possessing multiple required properties, particularly including binding affinities to specified protein targets.
Results
In this section, we first conduct the experiments on a benchmark to demonstrate ParetoDrug’s remarkable ability to generate molecules with multiple desired properties including the binding affinity and drug-like properties when compared with various baselines. Meanwhile, we also give the statistical analysis of the generated molecules of ParetoDrug. Then we use ParetoDrug to perform the case studies for the multi-objective target-aware drug discovery task, multi-target drug discovery task, and multi-target multi-objective drug discovery task respectively. In these case studies, ParetoDrug is able to generate the Pareto Dominate molecules over the known drug ligands in terms of the specified molecule property objectives, which exhibits the promising molecule discovery potential of ParetoDrug.
Benchmark experiments
In the benchmark experiments, we follow the settings as Qian et al.15 where there are 100 protein targets sampled from the public database of protein-ligand pairs BindingDB31 as the test set. For each test protein target, we generate 10 candidate molecules for evaluation. All 1000 candidate molecules are evaluated by a set of molecule property metrics, and the scores are averaged for an overall comparison. Please refer to Supplementary Information A and B for a detailed experimental and hyperparameter setup. We use several important metrics to evaluate the generated molecules, including docking score, uniqueness, LogP, QED, SA score, and NP-likeness described as follows.
- Docking score. Binding energy is regarded as a general indicator to describe the binding affinity between molecule ligands and target proteins. Specifically, we utilize a free and widely used tool called smina32 to compute the binding affinity. We use the negative value of the output by smina as the docking score. The higher the docking score is, the better the molecule is docked into the target protein.
- Uniqueness. Drug design models should be able to generate different molecules conditioning on different target proteins. The higher the uniqueness value is, the more sensitive the model is to the specified target protein. This metric is computed as follows:
-
(1)
where Sp indicates the set of test proteins, MSp denotes the collection of generated molecules for the target protein Sp∈Sp, # counts the number of molecules, and Set is an operator to remove the repeated molecules in the given set.
- LogP. A large LogP value indicates the substance is lipophilic, while a small LogP value means it is easy to dissolve in water. According to Ghose filter33, the LogP value of a druggable molecule should range from −0.4 to +5.6.
- QED. This score measures the drug-likeness and ranges from 0 to 1. A higher QED score indicates that a molecule is more likely to be a potential drug-like compound, with the desired molecular properties such as hydrogen bond acceptor, hydrogen bond donor, and polar molecular surface area34.
- SA score. The synthetic accessibility (SA) score indicates how difficult one molecule is to synthesize, which is calculated based on a combination of fragment contributions and a complexity penalty35. The range of the estimated SA metric is from 1 (easy to make) to 10 (very difficult to make).
- NP-likeness. Natural products play an important role in the history of drug discovery. Many drugs are natural products and their derivatives. The higher the score is, the more likely the molecule is to be a natural product. The calculated NP-likeness is typically in the range from -5 to 536.
The reported results of “Known ligands”, SBMolGen, LiGANN, SBDD-3D, and BeamLmser are from AlphaDrug15. The “Known ligands” indicates the original molecules binding to protein targets in the database. The results of LiGANN10 were collected on the web-based application provided in the original paper. SBMolGen37 is developed from ChemTS38 for target-specific molecular generation. The results of SBDD-3D18 were based on the released codes and trained model published by the authors. BeamLmser applies the beam search on the pretrained Lmser Transformer15. The beam size of BeamLmser is set at 10 to collect 10 molecules for each test protein target. Besides the above representative baselines, we also test three recent advanced methods. The first is Pocket2Mol20, which uses the equivariant generative network and autoregressive sampling scheme to generate three-dimensional molecules. For Pocket2Mol, we utilize the official codes and trained model for sampling molecules. The second is TargetDiff12, which develops a three-dimensional equivariant diffusion model to sample molecules. For TargetDiff, we also use the officially released trained model and codes for sampling. We keep the sample numbers of Pocket2Mol and TargetDiff at 100 for each test protein, which is the default configuration to ensure the quality of generated molecules. To make a fair comparison with other methods, for each test protein target, we randomly select 10 molecules from the generated 100 molecules of Pocket2Mol and TargetDiff for the evaluation. The third is CProMG28, which proposes a multi-constraint autoregressive model to generate small molecules with controllable properties. We use the official codes and default configurations of CProMG to generate 10 molecules for each test protein with the pretrained CProMG-VQSLT model, which is trained to control multiple property metrics including the docking score, LogP, QED, and SA score that are evaluated here.
Besides the above basic generative models, there also emerges another kind of fundamental approach that integrates the powerful MCTS-based searching technique to better control the molecule generation procedure of the pretrained autoregressive generative models with the simulation feedback, and AlphaDrug and the proposed ParetoDrug fall into this kind. For AlphaDrug15 which utilizes MCTS with the pretrained Lmser Transformer model to generate molecules based on given protein targets, we run the official codes and set iteration times (IT) at 150 when selecting the next atom symbol in MCTS. For ParetoDrug which conducts Pareto MCTS with the same pretrained Lmser Transformer model, we also set IT at 150 and let it optimize all objectives (docking score, LogP, QED, SA score, and NP-likeness) synchronously except the unoptimizable Uniqueness, which is a statistic metric for all generated molecules. In addition, we set the metric value of LogP as 1 if the molecule’s LogP value is in the range of [ − 0.4, 5.6], and 0 otherwise. After each Pareto MCTS, ParetoDrug obtains a global pool of Pareto optimal molecules. We choose the molecule with the largest reward vector summation value from the pool, which means this molecule has top rankings in each property metric. When testing, we collect 10 generated molecules for each test protein target from AlphaDrug and ParetoDrug.
Additionally, we compare a multi-objective drug discovery algorithm REINVENT 439 while its generation model is not conditioned on the protein information. It uses a reinforcement learning algorithm to generate optimized molecules compliant with a user-defined property profile defined as a multi-component score. We let REINVENT 4 optimize the docking score, LogP, QED, SA, and NP while setting their weights in the property profile all at 0.2. For each test protein target, we collected 10 molecules with the highest multi-component scores during the training process of REINVENT 4.
The results are shown in Table 1 and the direction of the arrow in the table means a better property score. The 95% confidence intervals for property scores of RL/MCTS are included. As we see, in terms of the docking score, ParetoDrug demonstrates superiority over all baselines except AlphaDrug. However, AlphaDrug is a single-objective target-aware drug discovery method that only optimizes the binding affinity. As AlphaDrug and ParetoDrug have the same iteration budgets (IT=150) for each atom symbol in sequence but ParetoDrug needs to optimize multiple objectives including the binding affinity, it is expected that ParetoDrug has a lower docking score than AlphaDrug. Meanwhile, although the docking score of ParetoDrug decreases slightly, other metrics including QED, SA score, and NP-likeness are improved significantly compared with AlphaDrug. Notably, QED changes from 0.4 to 0.6 (50% improvement) while NP-likeness changes from -0.9 to -0.4 (55.6% improvement). For the special LogP metric, although the average LogP value of AlphaDrug falls into the druggable molecule range, only 52.7% generated molecules of AlphaDrug satisfy the LogP range constraint if tested individually. On the contrary, 96.5% (83.1% improvement over AlphaDrug) generated molecules of ParetoDrug satisfy the LogP range constraint. These impressive results demonstrate that ParetoDrug is able to address the multi-objective target-aware drug discovery task by discovering novel compounds that possess multiple satisfactory properties including the binding affinity. On the other hand, we observe that the pretrained autoregressive Lmser Transformer with beam search (BeamLmser) cannot generate molecules with higher docking scores than the most recent TargetDiff. But with MCTS replacing beam search, AlphaDrug greatly boosts Lmser Transformer’s performance to find molecules with stronger binding affinity than BeamLmser even with the same docking time budgets15. Furthermore, ParetoDrug proposes the multi-objective Pareto MCTS to replace the MCTS used in AlphaDrug. With the same iteration times, ParetoDrug significantly improves multiple molecule properties compared with AlphaDrug while maintaining the docking score at the same level. Additionally, when compared with the multi-constraint conditional generation method CProMG, ParetoDrug has advantages in docking score, Uniqueness, SA score, and NP-likeness. In addition, the Uniqueness of CProMG is only 26.9% as it generates the same molecules for different protein targets, which is undesirable in de novo target-aware drug discovery tasks. Lastly, for REINVENT 4 which does not belong to the kind of target-aware drug discovery methods, we could see although it achieves superior performance in some metrics such as QED and NP, its docking score is much lower than ParetoDrug as it does not encode the protein-ligand prior to its generation model. This also indicates the importance of incorporating the protein target information into the molecule generation process as in the generative target-aware drug discovery methods.